Projects
Synaptic and intrinsic plasticity
The ability of our brains to learn rests on the plasticity of synapses. Our goal is to reach a gap-free understanding of learning from molecules to behavior. To achieve this goal, we work with genetically modified mice that we study at the levels of molecules, synapses, neural circuits and behavioral output.
We examine forms of synaptic plasticity—both long-term potentiation (LTP) and long-term depression (LTD)—in cerebellar slice preparations from rat and mouse brains. We have shown that molecular pathways involved in LTP and LTD at cerebellar synapses are very different from their hippocampal counterparts. LTD requires larger calcium transients than LTP, and depends on the activation of NMDA receptors and CaMKII / PKC, while LTP depends on phosphatase activation.
We also study Purkinje cell intrinsic plasticity, which is mediated by a downregulation of calcium-activated SK2 channels. These channels regulate dendritic signal processing and calcium transients, and modulate the spike output of Purkinje cells. We study these phenomena in slices, and examine the consequences of excitability changes using in vivo patch-clamp recordings.
Dendrites
Neurons receive most synaptic input on their dendrites, where incoming signals are processed and ultimately propagate towards the soma. It is not well understood how dendrites operate and how synaptic input is modulated while passing through the dendritic tree.
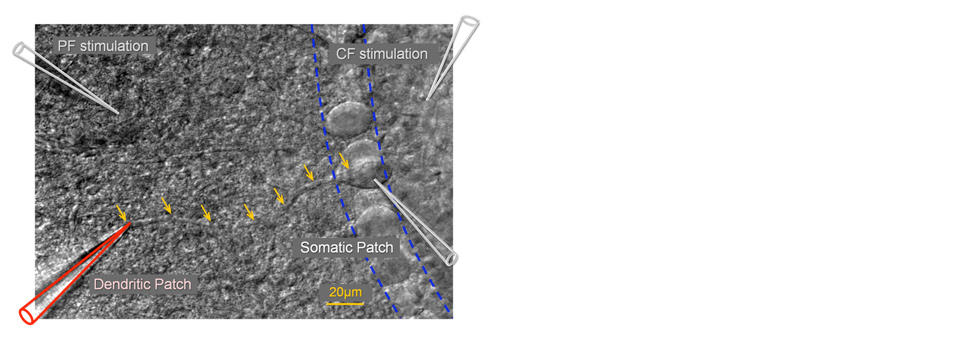
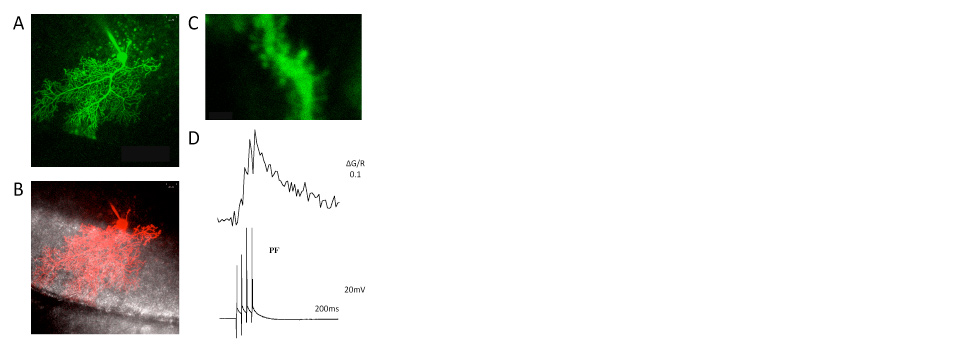
Using double- and triple-patch recordings from the soma and dendrite of Purkinje cells, we study dendritic signal processing. We discovered a novel form of dendritic plasticity, in which a modulation of SK2 channels alters dendritic excitability, controls the amplitude of local calcium transients, and changes the LTP / LTD probability. These excitability changes can be restricted to individual dendritic branches and may adjust the coupling strength to the primary dendrite.
We use confocal laser scan microscopy to monitor calcium transients in dendritic spines and to assess their role in synaptic plasticity. We study how sliding calcium amplitude thresholds interact with instructive signals to control LTP and LTD induction.
Autism
Autism Spectrum Disorder (ASD) is defined by two characteristic symptoms, deficits in social interaction and repetitive behaviors. An additional, often overlooked feature of autism is motor impairment, which is seen in ~80% of children with ASD. Reported motor problems include difficulties with gaze and eye movement control, pointing towards a cerebellar role in autism.
We study cerebellar abnormalities as well as motor coordination and learning in a mouse model for the human 15q11-13 duplication, which is one of the most frequent, but also one of the most penetrant genetic aberrations in autism.
In these mice, which show autism-resembling alterations in social behaviors, motor learning is affected and LTD at cerebellar synapses is impaired. Remarkably, similar LTD deficits have been described across various brain areas in different ASD mouse models. We study the implications of these LTD deficits for the cerebellar control of movements on the one hand, and for neural circuit development and function throughout the brain on the other.
